Using CellPipeline¶
A cell type specific analysis and visualization tool for the gene of interest¶
This notebook is built to be run automatically, you can just “Run All” cells. Beware: this requires some patience and high computational resources at the moment.
[286]:
import sys, os
sys.path.append(os.path.dirname(sys.path[0]))
from importlib import reload
import genereporter.cell_pipeline as cp
reload(cp)
cp = cp.CellPipeline(wdir = os.path.dirname(sys.path[0]), data_file="data/output/adata.h5ad") # instantiate the CellPipeline class with example data from scanpy
# set your gene of interest
GOI = "CASP8"
# set your cell type of interest
CELL_TYPE = 'T Cell'
# set the name of the column in adata.obs that contains your cell type annotations
CELL_TYPE_COL = 'manual_celltype_annotation'
[240]:
cp.adata
[240]:
AnnData object with n_obs × n_vars = 5397 × 16719
obs: 'sampleID', 'barcode', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'outlier', 'mt_outlier', '_scvi_batch', '_scvi_labels', 'leiden_res0_6', 'manual_celltype_annotation', 'celltypist_cell_label', 'celltypist_conf_score', 'celltypist_cell_label_coarse'
var: 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: '_scvi_manager_uuid', '_scvi_uuid', 'celltypist_cell_label_coarse_colors', 'celltypist_cell_label_colors', 'hvg', 'leiden', 'leiden_res0_6_colors', 'neighbors', 'pca', 'sampleID_colors', 'umap', 'manual_celltype_annotation_colors'
obsm: 'X_pca', 'X_scVI', 'X_umap'
varm: 'PCs'
layers: 'int_norm', 'log_int_norm', 'log_norm', 'norm', 'raw'
obsp: 'connectivities', 'distances'
Below is a list of possible coarse (level 1) cell types. Choose one of these as your cell type of interest above (cell_type = ‘[your cell type]’) to run the notebook automatically. Of course, you can rerun certain outputs on different cell types as well.
[232]:
# print cell type names here; easier to select
print(f"Cell types: ")
for cell_type in cp.adata.obs[CELL_TYPE_COL].unique():
print(f"\t{str(cell_type)}")
Cell types:
Monocyte / Dentritic Cell
T Cell
Endothelial Cell / Macrophage
Enterocyte Cells
Epithelial Cells / Goblet Cells
T Cell / Tuft Cells
Pericyte / Fibroblast
Gland Cells
Enteroendocrine Cells
Plasma B Cell
[234]:
# UMAP of coarse cell types
cp.plot_umap(color=CELL_TYPE_COL, legend_loc = "right margin")

Next, we provide a quick summary of the GOI’s expression class and mean expression level across all cell types.
[265]:
expr_sum = cp.explain_expr_celltypes(GOI=GOI, cell_type_col=CELL_TYPE_COL, layer='log_norm')
expr_sum
[265]:
| Cell type | Expression class | Avg. expression over cell type | |
|---|---|---|---|
| CASP8 | Epithelial Cells / Goblet Cells | very low | 0.291 |
| CASP8 | Enteroendocrine Cells | very low | 0.264 |
| CASP8 | Endothelial Cell / Macrophage | very low | 0.160 |
| CASP8 | T Cell | very low | 0.134 |
| CASP8 | Pericyte / Fibroblast | very low | 0.133 |
| CASP8 | Enterocyte Cells | very low | 0.119 |
| CASP8 | Plasma B Cell | very low | 0.116 |
| CASP8 | T Cell / Tuft Cells | very low | 0.113 |
| CASP8 | Monocyte / Dentritic Cell | very low | 0.090 |
| CASP8 | Gland Cells | very low | 0.024 |
[264]:
cp.plot_expressions(GOI, cell_type='T Cell / Tuft Cells',
cell_type_col=CELL_TYPE_COL, show_summary=True, col='log1p(means)',
layer = 'log_norm')
# Can change show_summary=False to hide the textual summary of the expression classes (quantile thresholds and cell counts per category)
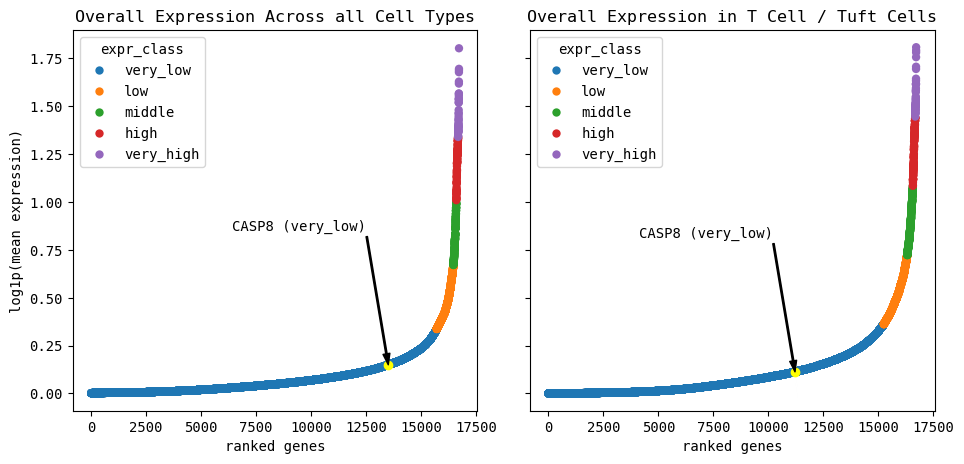
Summary for all cells:
Quantile thresholds:
very low: 93.941, low: 98.4748, middle: 99.2583, high: 99.7488, very high: 99.7500
Number of genes per category:
very_low: 15706
low: 758
middle: 131
high: 82
very_high: 42
Summary for T Cell / Tuft Cells cells:
Quantile thresholds:
very low: 91.2076, low: 97.5896, middle: 99.1088, high: 99.7488, very high: 99.7500
Number of genes per category:
very_low: 15249
low: 1067
middle: 254
high: 107
very_high: 42
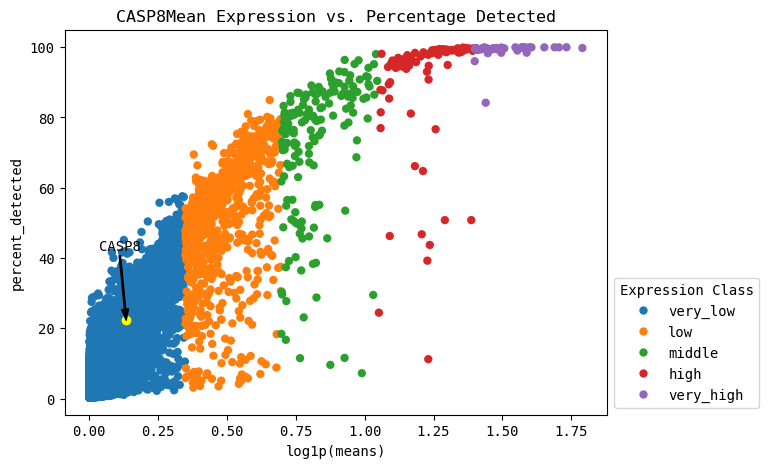
Expression vs. Detection visualization¶
This can contextualize the expression levels we observe in the standard scanpy plots. In single-cell RNA-seq, only a random sampling of the RNA present in a cell is selected to be sequenced. By pure chance, lowly expressed genes may not be present in all the sampled RNA due to their low prevalance. Here, we can inspect the maximum percentage of expression expected in all genes, specifically our gene of interest.
[273]:
cp.expression_vs_detection(GOI, cell_type_col=CELL_TYPE_COL, cell_type=CELL_TYPE)
# Can add (or remove) "cell_type=cell_type" to plot only the cell type of interest (or across all cell types)
# todo this section before dotplots etc.
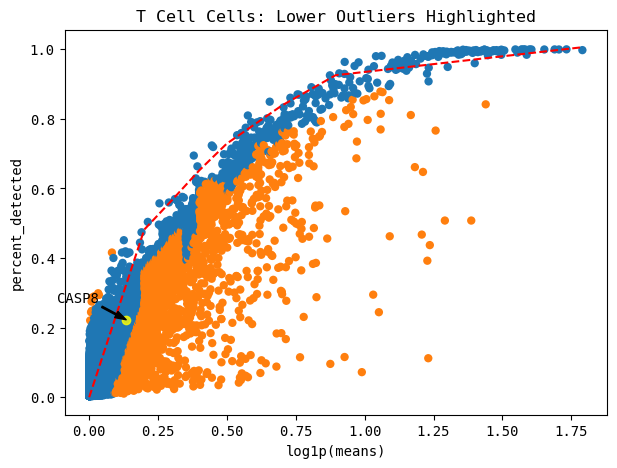
Automatically identify lower outliers (clue to look at celltype subset)¶
[307]:
cp.plot_outliers(GOI, outlier_threshold=0.08, s = 0.009, cell_type=CELL_TYPE, cell_type_col=CELL_TYPE_COL)
# Can add "cell_type=cell_type" to plot only the cell type of interest
This is how the maximum threshold curve approximation is calculated. This is primarily interesting for our fundamental understanding of the curve’s approximation through the spline’s 3rd derivative’s change points and the linear approximation of this curve.
[295]:
cp.fit_spline(plot=True, s = 0.009, cell_type=CELL_TYPE, cell_type_col=CELL_TYPE_COL)
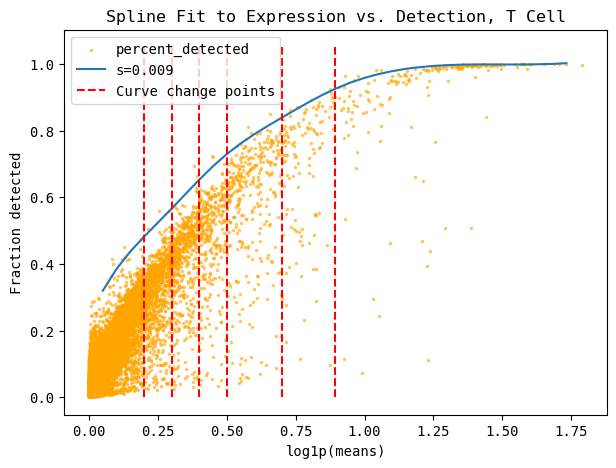
These are the top 5 number of outliers, sorted by their distance away from the maximum curve. You can show more or less by changing the head=n parameter.
[296]:
cp.list_outliers(cell_type=CELL_TYPE, cell_type_col=CELL_TYPE_COL)
# can show top n number of genes by adding "head=n"
[296]:
| log1p(means) | percent_detected | distance | is_outlier | |
|---|---|---|---|---|
| LYZ | 1.230811 | 0.112099 | 0.871155 | True |
| IL1B | 0.989631 | 0.072077 | 0.857988 | True |
| AIF1 | 0.926993 | 0.115620 | 0.802948 | True |
| CPVL | 0.621438 | 0.069668 | 0.732717 | True |
| LST1 | 0.765332 | 0.114879 | 0.730314 | True |
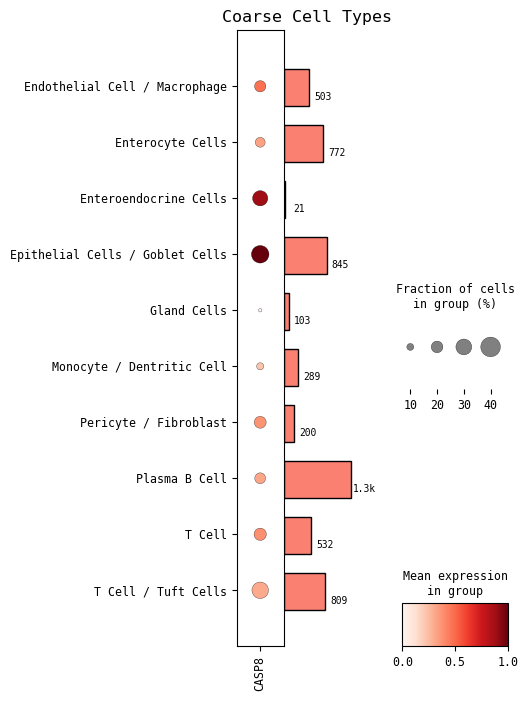
GOI expression across cell types¶
Now we show the standard scanpy plots of our GOI’s expression across both coarse cell types and fine cell types. The fine cell type automatically shown in the one you set at the beginning of this notebook. You can rerun the cell with other cell types of interest by setting the cell_type=[‘your cell type’] parameter.
[297]:
# GOI expression across coarse cell types
cp.dotplot(GOI, cell_type_col=CELL_TYPE_COL)
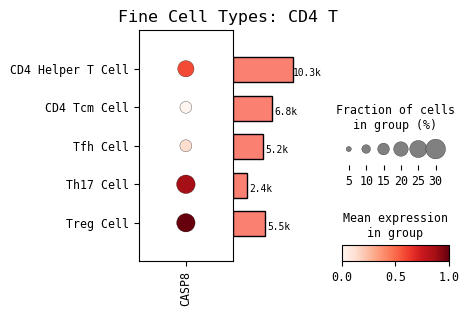
[17]:
# GOI expression in fine cell type
# this is sample output from a dataset that has both coarse and fine cell type annotations
cp.dotplot(GOI, cell_type=cell_type, cell_type_col=CELL_TYPE_COL)
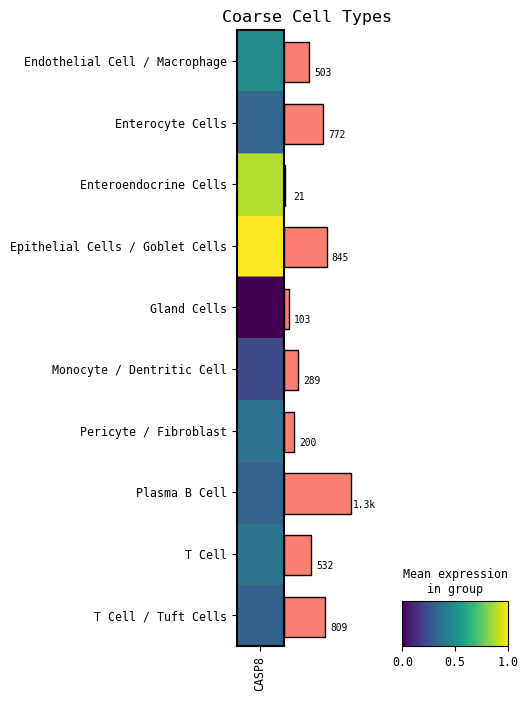
[306]:
# GOI expression across coarse cell types
# This is similar to the coarse cell type dotplot previously, just a different visualization
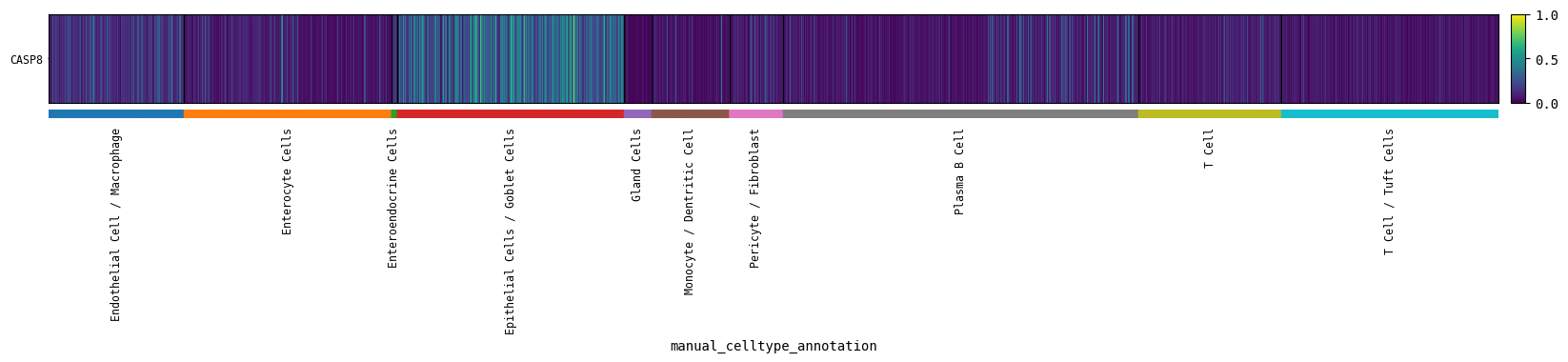
cp.matrixplot(GOI, cell_type_col=CELL_TYPE_COL)
[304]:
# GOI expression across coarse cell types
# Individual vertical "lines" correspond to individual cells
# A more fine grained visual than the mean expression plots shown before
cp.heatmap(GOI, cell_type_col=CELL_TYPE_COL, layer="log_int_norm")